

ヒトとウイルス間のタンパク質相互作用を正確に予測する深層学習モデルを開発-新型コロナウイルスなどへの抗ウイルス薬のコンピュータ支援設計を加速-
プレス情報 掲載日:2021年06月25日
ヒトとウイルス間のタンパク質相互作用を
正確に予測する深層学習モデルを開発
― 新型コロナウイルスなどへの抗ウイルス薬のコンピュータ支援設計を加速 ―
九州工業大学大学院情報工学研究院の倉田博之教授が研究代表者を務める研究グループは、独自に開発した深層学習モデルを用いて、タンパク質のアミノ酸配列情報のみから世界最高レベルの正答率でヒトとウイルス間のタンパク質相互作用(Human-Virus Protein-Protein Interaction(HV-PPI))を予測することに成功しました(図1)。従来、タンパク質のアミノ酸配列情報のみを用いたHV-PPIの予測は困難な課題と考えられていましたが、本研究グループはタンパク質のアミノ酸配列を文脈の流れと捉えて解析することで、この困難な課題を解決しました。ウイルスの感染にはHV-PPIが決定的な役割を果たすため、今後、本研究グループが開発した深層学習モデルは、新型コロナウイルスを含むウイルス群とヒトのタンパク質相互作用を解明し、抗ウイルス治療薬の開発を加速することが期待されます。
ポイント
- ヒトとウイルスのタンパク質アミノ酸配列情報(ゲノム情報)のみから、ヒトとウイルス間のタンパク質相互作用(HV-PPI)を正確に予測する深層学習モデルを開発した。
- 自然言語の文脈の流れと意味を解析する最新のAI技術をタンパク質アミノ酸配列の解析に応用する画期的な技術を提案した。
- 開発した深層学習モデルにより、新型コロナウイルスやその変異型を含むウイルス群に対する治療薬の開発を加速することが期待される。
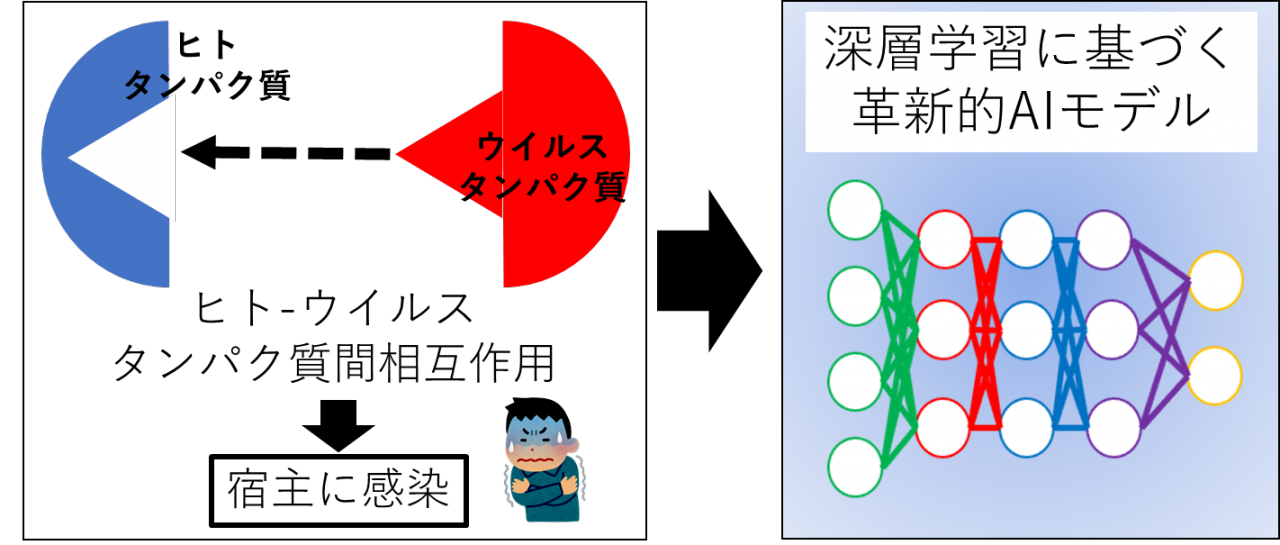
図1 ヒトとウイルスのタンパク質間相互作用を予測する深層学習モデルの開発
現在、全世界で300万人以上の死者を出している新型コロナウイルス感染症(Coronavirus disease 2019(COVID-19))の原因であるSARSコロナウイルス2(Severe acute respiratory syndrome coronavirus 2(SARS-CoV-2))には、既存薬による治療効果は少ないため、新しい抗ウイルス薬の開発が焦眉の急です。ウイルスは、ヒト(宿主)細胞表面上の特定のタンパク質と相互作用し、細胞内に侵入します。侵入後ウイルスは、ヒトの転写・翻訳・ゲノム複製などに関わるタンパク質をハイジャックしながら増殖し、ヒト細胞を死滅させます。すなわち、ヒトとウイルス間のタンパク質相互作用(HV-PPI)が、ウイルス感染症では決定的な役割を果たします。よって、HV-PPIを同定することは、ウイルスの感染メカニズムを解明して、抗ウイルス薬(HV-PPI阻害剤)の標的を発見することにつながります。
HV-PPI同定のための生物学実験は膨大な時間と費用がかかるため、コンピュータによるHV-PPIの予測が期待されています。従来のHV-PPIのコンピュータ予測法は、タンパク質立体構造情報に基づく分子動力学法を用いたドッキングシミュレーションです。分子動力学法は、タンパク質の鍵と鍵穴の形状を調べて、鍵が鍵穴にはまる条件をコンピュータシミュレーションによって見つける方法です(図1)。しかし、立体構造情報を解明することは難しく、ウイルスやヒトのタンパク質立体構造は不明なものが多いため、分子動力学法のHV-PPI予測への応用は限定的です。一方、ハイスループットな実験方法により、ヒトやウイルスのタンパク質のアミノ酸配列情報は容易に得ることができます。タンパク質立体構造情報を用いることなく、ゲノムの塩基配列情報(タンパク質のアミノ酸配列情報)のみからHV-PPIを予測できれば革新的です。しかし、タンパク質間相互作用特有の鍵と鍵穴の立体的構造を考慮せずに、アミノ酸配列情報だけを用いるHV-PPI予測法は、これまで困難な課題とみなされてきました。
本研究グループは、タンパク質のアミノ酸配列を文脈の流れと捉え、長鎖アミノ酸配列の順序パターンから鍵と鍵穴の立体的特徴を抽出することで、この問題を克服できると考えました。そこで、このアイデアを実装するために、時系列データから未来を予測する深層学習モデルLong Short-Term Memory(LSTM)と自然言語における単語の分散表現技術word2vecを応用しました。その結果、LSTMとword2vecの両者を組み合わせて開発した深層学習モデルは、アミノ酸配列の文脈の流れから鍵と鍵穴の立体的特徴を的確に捉えて、正答率98%という世界最高レベルでHV-PPIを予測することに成功しました。
開発した深層学習モデルはLSTM-PHVと命名し、ウェブサイト において公開しています。LSTM-PHVは、タンパク質のアミノ酸配列情報があれば、SARS-CoV-2を含む多様なウイルス群や変異型ウイルス群とヒトのHV-PPIを予測できます。HV-PPIの正確な予測は、ウイルスの感染メカニズムを解明して、抗ウイルス薬の開発に貢献します。
なお、この研究成果は、Oxford University Press社の「Briefings in Bioinformatics」のオンライン版において2021年6月23日に公開されました。
■ 論文の詳細情報
| タイトル | “LSTM-PHV: Prediction of human-virus protein-protein interactions by LSTM with word2vec” |
| 著者名 | Sho Tsukiyama, Md Mehedi Hasan, Satoshi Fujii, Hiroyuki Kurata |
| 雑誌 | Briefings in Bioinformatics (Oxford University Press社) |
| DOI | 10.1093/bib/bbab228 |
※ 本研究は JSPS科研費19H04208 の助成を受けたものです。





















